Read and merge multiple files by folder
By Synnøve Yndestad in R RNAseq sequencing data vrangeling
November 30, 2022
Reading multiple files by folder
Often the data we need is spread out across multiple files, and we need a way to read all the files and merge the content.
The goal is to generate a tidy data frame.
Tidy data have variables in columns and observations in rows. Here I demonstrate how to gather data from multiple files into a tidy dataset from;
1- A folder with one file pr measurement.
2- A folder where you have one file pr sample with multiple measurements.
3- A folder using regex to select specific files.
4- Show off some superpowers by applying this in creating a function to merge a folder of VCF files into one long data frame.
Files used in this “How to” can be downloded from here:
https://github.com/Syndestad/Learning-curve
Load the necessary libraries:
library(tidyverse)
library(fs)
library(here)
The fs package provides a cross-platform, uniform interface to file system operations. It is very useful when working with file-paths. The here package is very useful for setting your file path relative to here::here(). Setting a relative path to here::here() will make your code transportable, and everything will work even of you move the script file to another location or computer.
Where are we?
here::here()
## [1] "/Users/synnoveyndestad/Syndestad.github.io"
1- A folder with one file pr measurement
We have a folder of files that has samples in rows, and observations in columns with the following structure:

Bcells_ave <- read_csv("OneFilePrMeasurement/Bcells.ave.csv")
head(Bcells_ave)
## # A tibble: 6 × 2
## SampleId Bcells
## <dbl> <dbl>
## 1 1 -0.0909
## 2 2 -1.00
## 3 3 -1.00
## 4 4 0.818
## 5 5 1.73
## 6 6 -1.00
In stead of reading in files one by one, we can use fs::dir_map().
dir_map(path, function)
dir_map(), applies a function to each entry in the path and returns the result in a list.
Set the file path by naming the folder in your working directory where your data is, and pasting it to here::here()
Select appropriate function for file type, i.e use read_csv for csv files, readxl::read_excel() for excel files.
Then, if your data contains a key that is identical in each file such as “SampleId”, you can merge the list of files by calling full_join within Reduce.
# Set the name of the folder to read in your working directory.
MyFolder = "/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrMeasurement"
here::here()
## [1] "/Users/synnoveyndestad/Syndestad.github.io"
# Read files
ListOfFiles = fs::dir_map( paste0(here::here(), MyFolder), read_csv)
# Merge
MergedFiles = Reduce(full_join,ListOfFiles)
TADAAAA!
Yes, it really is that simple.
The list of files:
 The merged data frame:
The merged data frame:
MergedFiles %>% knitr::kable()
| SampleId | Bcells | Chemokine12 | Dendritic | MastC | Tcells |
|---|---|---|---|---|---|
| 1 | -0.0908674 | -0.6123724 | -1.4661469 | 1.6035675 | -1.38873 |
| 2 | -0.9995412 | 1.2247449 | 1.1211712 | -0.8017837 | 1.38873 |
| 3 | -0.9995412 | 0.0000000 | -0.6037076 | -1.6035675 | 0.46291 |
| 4 | 0.8178064 | 0.6123724 | 0.2587318 | -0.8017837 | 0.46291 |
| 5 | 1.7264802 | 1.2247449 | 0.2587318 | 0.0000000 | -0.46291 |
| 6 | -0.9995412 | 1.2247449 | -0.6037076 | -0.8017837 | -1.38873 |
| 7 | -0.9995412 | -1.2247449 | 1.9836105 | 0.0000000 | 0.46291 |
| 8 | 0.8178064 | -0.6123724 | 0.2587318 | 0.8017837 | -0.46291 |
| 9 | 0.8178064 | -1.2247449 | -0.6037076 | 0.8017837 | -0.46291 |
| 10 | -0.0908674 | -0.6123724 | -0.6037076 | 0.8017837 | 1.38873 |
Note:
as.data.frame(ListOfFiles) works surprisingly well too, but does not merge by key.
as.data.frame(ListOfFiles) %>% knitr::kable()
| SampleId | Bcells | SampleId.1 | Chemokine12 | SampleId.2 | Dendritic | SampleId.3 | MastC | SampleId.4 | Tcells |
|---|---|---|---|---|---|---|---|---|---|
| 1 | -0.0908674 | 1 | -0.6123724 | 1 | -1.4661469 | 1 | 1.6035675 | 1 | -1.38873 |
| 2 | -0.9995412 | 2 | 1.2247449 | 2 | 1.1211712 | 2 | -0.8017837 | 2 | 1.38873 |
| 3 | -0.9995412 | 3 | 0.0000000 | 3 | -0.6037076 | 3 | -1.6035675 | 3 | 0.46291 |
| 4 | 0.8178064 | 4 | 0.6123724 | 4 | 0.2587318 | 4 | -0.8017837 | 4 | 0.46291 |
| 5 | 1.7264802 | 5 | 1.2247449 | 5 | 0.2587318 | 5 | 0.0000000 | 5 | -0.46291 |
| 6 | -0.9995412 | 6 | 1.2247449 | 6 | -0.6037076 | 6 | -0.8017837 | 6 | -1.38873 |
| 7 | -0.9995412 | 7 | -1.2247449 | 7 | 1.9836105 | 7 | 0.0000000 | 7 | 0.46291 |
| 8 | 0.8178064 | 8 | -0.6123724 | 8 | 0.2587318 | 8 | 0.8017837 | 8 | -0.46291 |
| 9 | 0.8178064 | 9 | -1.2247449 | 9 | -0.6037076 | 9 | 0.8017837 | 9 | -0.46291 |
| 10 | -0.0908674 | 10 | -0.6123724 | 10 | -0.6037076 | 10 | 0.8017837 | 10 | 1.38873 |
2- One file pr sample with multiple measurements
When you have one file pr sample and need to keep track of the file name as sample name, we can use fs::dir_ls().
dir_ls() is equivalent to the ls command. It returns filenames as a named fs_path character vector.
Consider the following file structure:
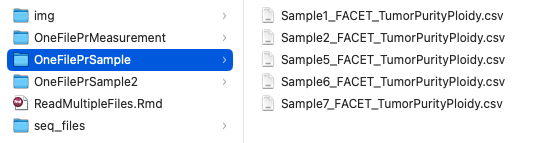
read_csv("OneFilePrSample/Sample1_FACET_TumorPurityPloidy.csv")
## # A tibble: 1 × 2
## purity ploidy
## <dbl> <dbl>
## 1 0.0579 4.30
To read and merge all files in the folder, list all paths in the folder with dir_ls().
MyFolder = "OneFilePrSample"
files = fs::dir_ls(MyFolder)
files
## OneFilePrSample/Sample1_FACET_TumorPurityPloidy.csv
## OneFilePrSample/Sample2_FACET_TumorPurityPloidy.csv
## OneFilePrSample/Sample5_FACET_TumorPurityPloidy.csv
## OneFilePrSample/Sample6_FACET_TumorPurityPloidy.csv
## OneFilePrSample/Sample7_FACET_TumorPurityPloidy.csv
Read as a large data frame using an appropriate function.
Use read_csv for csv files, read_xls for excel files etc.
Set name of file with th “.id” argument.
allFiles = files %>% map_df(read_csv, .id = "filename")
allFiles %>% knitr::kable()
| filename | purity | ploidy |
|---|---|---|
| OneFilePrSample/Sample1_FACET_TumorPurityPloidy.csv | 0.0579344 | 4.300485 |
| OneFilePrSample/Sample2_FACET_TumorPurityPloidy.csv | NA | 2.000000 |
| OneFilePrSample/Sample5_FACET_TumorPurityPloidy.csv | 0.3919855 | 2.121393 |
| OneFilePrSample/Sample6_FACET_TumorPurityPloidy.csv | NA | 2.000000 |
| OneFilePrSample/Sample7_FACET_TumorPurityPloidy.csv | 0.9386437 | 2.289907 |
And we have successfully merged the files to a tidy data frame.
The Sample names may need some editing.
Remove file path and file-extension from sample name, and the merged data frame is ready:
# Add Sample ID column
allFiles$SampleID = allFiles$filename
# reorder columns
allFiles = allFiles %>% select(filename, SampleID, everything())
# Remove folder name
allFiles$SampleID <- str_remove(allFiles$SampleID, paste0(MyFolder, "/"))
# Remove all after "_"
allFiles$SampleID <- gsub("_.*","",allFiles$SampleID)
head(allFiles) %>% knitr::kable()
| filename | SampleID | purity | ploidy |
|---|---|---|---|
| OneFilePrSample/Sample1_FACET_TumorPurityPloidy.csv | Sample1 | 0.0579344 | 4.300485 |
| OneFilePrSample/Sample2_FACET_TumorPurityPloidy.csv | Sample2 | NA | 2.000000 |
| OneFilePrSample/Sample5_FACET_TumorPurityPloidy.csv | Sample5 | 0.3919855 | 2.121393 |
| OneFilePrSample/Sample6_FACET_TumorPurityPloidy.csv | Sample6 | NA | 2.000000 |
| OneFilePrSample/Sample7_FACET_TumorPurityPloidy.csv | Sample7 | 0.9386437 | 2.289907 |
The merged data frame with the added and cleaned sample names are ready!
3- Read multiple files based on regexp, create count matrix from RNAseq
Often, you have a mix of files in a folder. Here, we have RNAseq output by transcript (.quant.sf) and gene (.quant.genes.sf) all in the same folder. To make a count matrix for downstream analysis we only want to read the files including genes, the “quant.genes.sf” file extension and not the transcript with the “quant.sf” extension.
Then we can use regular expressions (regex) in the read call to select only the files that we want.
More on regular expressions here:
https://www.rexegg.com/regex-quickstart.html
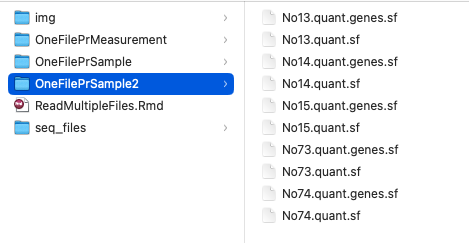
The goal is to read only the “quant.genes.sf” files in the folder and make a count matrix.
List all paths in the folder that has a file name that ends with “.genes.sf” by using “*” in the regexp argument:
/Users/syndestad/Documents/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/
MyFolder = "/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/"
# List filepaths of files containig a spesific expression
files = fs::dir_ls(paste0(here::here(), MyFolder), regexp = "*.genes.sf")
files
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/No13.quant.genes.sf
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/No14.quant.genes.sf
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/No15.quant.genes.sf
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/No73.quant.genes.sf
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/No74.quant.genes.sf
Read as a large data frame using an appropriate function i.e read.table for tabular data,
read_csv for csv files, read_xls for excel files.
Add name of file with the “.id” function.
allFiles = files %>% map_df(read.table, .id = "SampleID", header = TRUE)
head(allFiles) %>% knitr::kable()
| SampleID | Name | Length | EffectiveLength | TPM | NumReads | |
|---|---|---|---|---|---|---|
| 1…1 | /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/No13.quant.genes.sf | ENSG00000121879.6 | 3620 | 2828.85 | 12.489 | 1770.99 |
| 2…2 | /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/No13.quant.genes.sf | ENSG00000091831.24 | 6194 | 5736.83 | 0.598 | 172.00 |
| 3…3 | /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/No13.quant.genes.sf | ENSG00000171862.11 | 2573 | 1773.48 | 50.906 | 4525.55 |
| 4…4 | /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/No13.quant.genes.sf | ENSG00000139618.17 | 7505 | 6155.61 | 3.299 | 1018.00 |
| 5…5 | /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/No13.quant.genes.sf | ENSG00000142208.18 | 2681 | 2506.42 | 25.740 | 3234.00 |
| 6…6 | /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/OneFilePrSample2/No13.quant.genes.sf | ENSG00000141510.18 | 1437 | 1352.28 | 16.036 | 1087.00 |
Now we have all the files in a Very Long format.
Clean up the sample names by removing file-path and file-extension from sample name.
# Remove filepath
allFiles$SampleID <- str_remove(allFiles$SampleID, paste0(here::here(), MyFolder))
# Remove file-extension
allFiles$SampleID <- str_remove(allFiles$SampleID, ".quant.genes.sf")
head(allFiles, n= 15) %>% knitr::kable()
| SampleID | Name | Length | EffectiveLength | TPM | NumReads | |
|---|---|---|---|---|---|---|
| 1…1 | No13 | ENSG00000121879.6 | 3620 | 2828.85 | 12.489 | 1770.99 |
| 2…2 | No13 | ENSG00000091831.24 | 6194 | 5736.83 | 0.598 | 172.00 |
| 3…3 | No13 | ENSG00000171862.11 | 2573 | 1773.48 | 50.906 | 4525.55 |
| 4…4 | No13 | ENSG00000139618.17 | 7505 | 6155.61 | 3.299 | 1018.00 |
| 5…5 | No13 | ENSG00000142208.18 | 2681 | 2506.42 | 25.740 | 3234.00 |
| 6…6 | No13 | ENSG00000141510.18 | 1437 | 1352.28 | 16.036 | 1087.00 |
| 7…7 | No13 | ENSG00000012048.23 | 2875 | 2646.37 | 1.621 | 215.00 |
| 1…8 | No14 | ENSG00000121879.6 | 4203 | 3192.80 | 16.732 | 1529.00 |
| 2…9 | No14 | ENSG00000091831.24 | 5318 | 4864.04 | 3.513 | 489.00 |
| 3…10 | No14 | ENSG00000171862.11 | 3639 | 2538.89 | 64.437 | 4682.38 |
| 4…11 | No14 | ENSG00000139618.17 | 5306 | 4250.20 | 2.252 | 274.00 |
| 5…12 | No14 | ENSG00000142208.18 | 2647 | 2545.31 | 44.915 | 3272.00 |
| 6…13 | No14 | ENSG00000141510.18 | 2396 | 2324.18 | 16.521 | 1099.00 |
| 7…14 | No14 | ENSG00000012048.23 | 5903 | 5439.87 | 1.182 | 184.00 |
| 1…15 | No15 | ENSG00000121879.6 | 3147 | 2626.14 | 11.999 | 1260.00 |
For the count matrix, we don’t need the “Length” or “EffectiveLength” columns.
Select columns to keep by using select().
Here, we want to use NumReads in the count matrix.
Pivot wider to create a count matrix with sample names in columns, and ENSEMBL ID (Name) as rows.
CountDF <- allFiles %>% select(SampleID, Name, NumReads) %>%
pivot_wider(names_from = SampleID,
values_from = NumReads) %>%
as.data.frame()
# Set rownames
MyRownames = CountDF$Name
rownames(CountDF) = MyRownames
# Remove names column and print matrix
MyMatrix = CountDF[, -1] %>% as.matrix()
MyMatrix %>% knitr::kable()
| No13 | No14 | No15 | No73 | No74 | |
|---|---|---|---|---|---|
| ENSG00000121879.6 | 1770.99 | 1529.00 | 1260.00 | 1167.00 | 1578.00 |
| ENSG00000091831.24 | 172.00 | 489.00 | 77.00 | 359.00 | 2043.00 |
| ENSG00000171862.11 | 4525.55 | 4682.38 | 2738.66 | 3992.53 | 4896.19 |
| ENSG00000139618.17 | 1018.00 | 274.00 | 1591.00 | 586.00 | 755.00 |
| ENSG00000142208.18 | 3234.00 | 3272.00 | 4380.00 | 4580.00 | 3475.00 |
| ENSG00000141510.18 | 1087.00 | 1099.00 | 1046.00 | 2738.00 | 1550.00 |
| ENSG00000012048.23 | 215.00 | 184.00 | 450.00 | 464.00 | 597.00 |
TADAAAA!
There is your count matrix ready to analyze!
4- Read and merge VCF files
Now that we have become a file-reading and merging wizard, lets try something more complex. Lets make a function that will read the FIX part of a VCF file, and use that when reading all the vcf files in a folder.
The Variant Call Format (VCF) stores the location and type of variant deviating from the reference genome.
The header starts with # and contains various metadata i.e what reference genome was used. The body has 8 mandatory columns, but can contain multiple others as well. We are only interested in the body, aka the FIX part in this exersice.
vcfR is an R package for working with vcf files, see details at:
https://knausb.github.io/vcfR_documentation/index.html
Load packages
library(tidyverse)
library(fs)
library(here)
library(vcfR)
We start with a folder containing a mix of files, vcf and the annotated csv file.

List all paths in the folder containing “*.vcf” to select only the .vcf files.
MyFolder = "/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/seq_files"
files = fs::dir_ls(paste0(here::here(), MyFolder),
regexp = "*.vcf")
files
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/seq_files/BT20_S8.vcf
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/seq_files/HCC1143_S6.vcf
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/seq_files/HCC1937_S3.vcf
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/seq_files/MB157_S2.vcf
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/seq_files/MB330_S3.vcf
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/seq_files/MB436_S7.vcf
## /Users/synnoveyndestad/Syndestad.github.io/content/blog/2022-11-30-read-and-merge-multiple-files-by-folder/seq_files/MCF-7_S6.vcf
Read as a large data frame using an appropriate function. But now, we have to MAKE the function to use.
We can read a vcf file with read.vcfR() and then use getFIX() to get the fixed/body info.
If we pipe them together, we get:
read.vcfR("seq_files/BT20_S8.vcf", verbose = FALSE ) %>%
getFIX(getINFO = TRUE) %>% as.data.frame() %>%
DT::datatable()
Make it into a function:
getVCFfix <- function (vcfFile) {
read.vcfR(vcfFile, verbose = FALSE ) %>%
getFIX(getINFO = TRUE) %>% as.data.frame()
}
Read all vcf files in the selected folder as a large data frame with the generated function getVCFfix().
Add the file-path-name with the “.id” argument so we keep track of which sample the data originates from.
## Read the FIX part of all vcf files in the folder using the generated function **getVCFfix**
allFiles = files %>% map_df(getVCFfix, .id = "SampleID")
# Clean up Sample ID, remove path names
allFiles$SampleID <- str_remove(allFiles$SampleID, paste0(here::here(), MyFolder, "/"))
# Remove everything after "_":
allFiles$SampleID <- sub("_[^_]+$", "", allFiles$SampleID)
allFiles %>% DT::datatable()
Tadaaa!
Now we have a Very Long data frame with all the FIX info from a folder of vcf files, with a column specifying SampleID.
Sweet!
# count all variants from each sample
table(allFiles$SampleID)
##
## BT20 HCC1143 HCC1937 MB157 MB330 MB436 MCF-7
## 681 732 698 803 780 791 728
- Posted on:
- November 30, 2022
- Length:
- 9 minute read, 1865 words
- Categories:
- R RNAseq sequencing data vrangeling