Calculate Z-Score and plot heatmaps
By Synnøve Yndestad in R RNAseq Statistics Visualizations
June 29, 2022
Z-score
Z-score is a measure for how values deviates from the mean in a given population.
A value with z-score = 0 is the average value in the data. Negative values is below average, and positive is above.
The formlua for calculating z-score is:
z-score = (x-μ)/σ
x = one observation in the population
μ = the mean of the population
σ = the standard deviation of the population
Calculating this in R can be done by:
library(tidyverse)
# Enter some data and calculate z-score
data <- c(1,5,9,12,14,23)
z_scores <- (data-mean(data))/sd(data)
z_scores
## [1] -1.2620630 -0.7398300 -0.2175971 0.1740777 0.4351941 1.6102183
The final number 23 is 1.6 standard deviations away from the mean.
mean(data)
## [1] 10.66667
Visualize how far above or below average the values are:
par(mfrow=c(1,2))
plot(data, type="b", ylab = "data value")
plot(z_scores, type="b", col="black", ylab = "z score")
abline(h = 0, col = "brown", lty = 4)
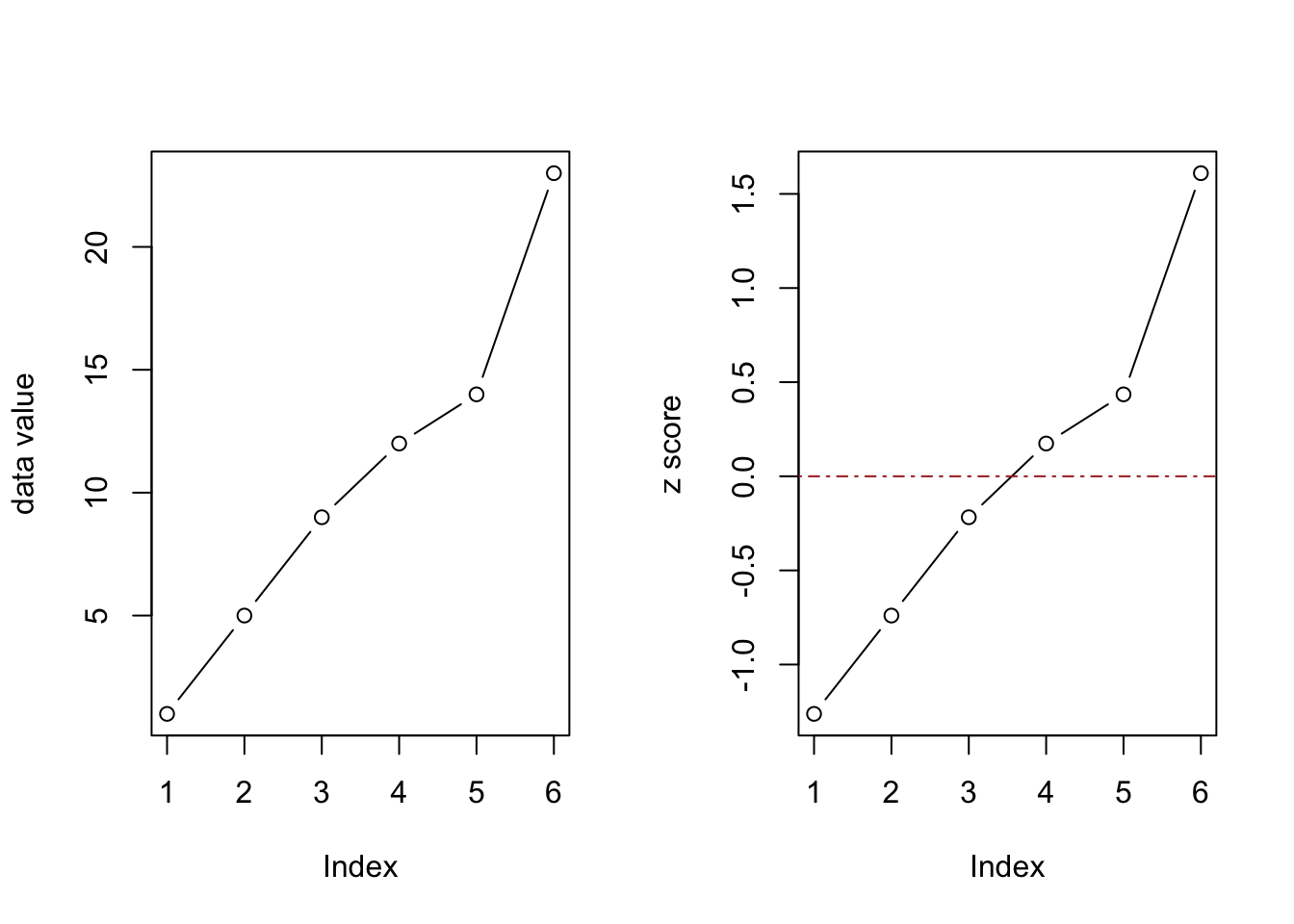
Calculating z-score is a handy way to standardize, or normalize data.
This is frequently used in gene expression studies to visualize heat maps of differential expressed genes.
The DE analysis is performed on normalized counts before log2 and Z-score transformation.
In R, scale() can be used for z-score transformation of large matrices.
Compared with manual Z-score calculation, we get identical results when using scale().
# Manually calculated z-score
z_scores
## [1] -1.2620630 -0.7398300 -0.2175971 0.1740777 0.4351941 1.6102183
# z-score using scale()
scale(data) %>% as.vector()
## [1] -1.2620630 -0.7398300 -0.2175971 0.1740777 0.4351941 1.6102183
Scaling is part of a normalization approach to be able to compare variance between samples.
scale() is performed column-wise.
In a matrix of gene expression data, the matrix must be transposed before and after z-score transformation if you want to compare gene expression between samples.
This will scale the expression of each gene compared to the mean of the expression in the population.
Doing scale on the columns will compare the expression of the gene compared to the other genes within the sample.
For genes in columns and samples in rows, use:
scale(data)
For genes in rows and samples in columns, use:
t(scale(t(data)))
Z-Score normalization with gene expression data:
Here is a demonstration of z-score normalization using gene expression from the airway dataset. The airway data set is available on Bioconductor.
Get the airway dataset
library(airway)
data(airway)
airway
## class: RangedSummarizedExperiment
## dim: 64102 8
## metadata(1): ''
## assays(1): counts
## rownames(64102): ENSG00000000003 ENSG00000000005 ... LRG_98 LRG_99
## rowData names(0):
## colnames(8): SRR1039508 SRR1039509 ... SRR1039520 SRR1039521
## colData names(9): SampleName cell ... Sample BioSample
Extract the count matrix
airway.matrix = assay(airway)
# matrix dimension before removing low count genes
dim(airway.matrix)
## [1] 64102 8
By removing low count genes, the mean variance can be estimated more reliable.
Therefore, genes with 0 counts should be removed for some applications.
# Remove genes with 0 counts
# Sett genes to keep, all genes that are not expressed to be removed
keep <- rowSums(airway.matrix) > 1
airway.matrix <- airway.matrix[keep,]
# Check dimensions
dim(airway.matrix)
## [1] 29391 8
Do log2 transform the count matrix to get normal distributed data before scaling and calculate z-score:
scale(log2(airway.matrix+1)) %>% head()
## SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516
## ENSG00000000003 1.1260901 1.0208946 1.15821433 1.0901426 1.2602939
## ENSG00000000419 0.9916230 1.0714161 1.03630532 1.0488761 1.0238041
## ENSG00000000457 0.7814606 0.7483022 0.72915287 0.7529099 0.7121244
## ENSG00000000460 0.2582945 0.2647380 0.06166199 0.1873347 0.3058427
## ENSG00000000938 -1.2212054 -1.1974507 -0.87556058 -1.1439277 -1.0090925
## ENSG00000000971 1.6893080 1.7850331 1.85914197 1.9600613 1.8952581
## SRR1039517 SRR1039520 SRR1039521
## ENSG00000000003 1.1574578 1.2089940 1.0829765
## ENSG00000000419 1.0631139 0.9851562 1.0406101
## ENSG00000000457 0.7558361 0.7730355 0.7564559
## ENSG00000000460 0.1806572 0.3666383 0.2817005
## ENSG00000000938 -1.2724024 -1.2215628 -1.1888027
## ENSG00000000971 1.9797589 1.9052498 2.0258340
Plotting heatmap
Plotting heatmap with and without log2 normalization, and also plotting when scaling with and without transposing the matrix is a nice visual demonstration of what happens to the data during z-score scaling.
Observe the difference in the heatmap when plotting the data with and without log2 normalization, and scaling with and without transposing the matrix.
# Get metadata for heatmap
metadata = as.data.frame(colData(airway))
# Extract genes with highest variance
topVarGenes <- order(-rowVars(assay(airway)))[0:50]
mat <- assay(airway)[ topVarGenes, ]
# The count matrix has samples in columns, and genes in rows
mat %>% head()
## SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516
## ENSG00000166923 126514 64824 156189 59216 247345
## ENSG00000115414 260262 213986 513766 273878 252667
## ENSG00000011465 219586 191767 422752 214062 293318
## ENSG00000198804 210138 213952 265361 169551 397791
## ENSG00000115461 74051 79611 328753 137165 241640
## ENSG00000087086 236805 138524 236361 95045 343728
## SRR1039517 SRR1039520 SRR1039521
## ENSG00000166923 336864 98504 56549
## ENSG00000115414 399841 345651 370450
## ENSG00000011465 313112 378834 372489
## ENSG00000198804 401539 233051 248032
## ENSG00000115461 216484 145858 148389
## ENSG00000087086 229323 167067 114257
Plot the heatmap using pheatmap() of un-normalized raw counts:
pheatmap::pheatmap(mat,
annotation = select(metadata, cell, dex),
main = "Raw counts")
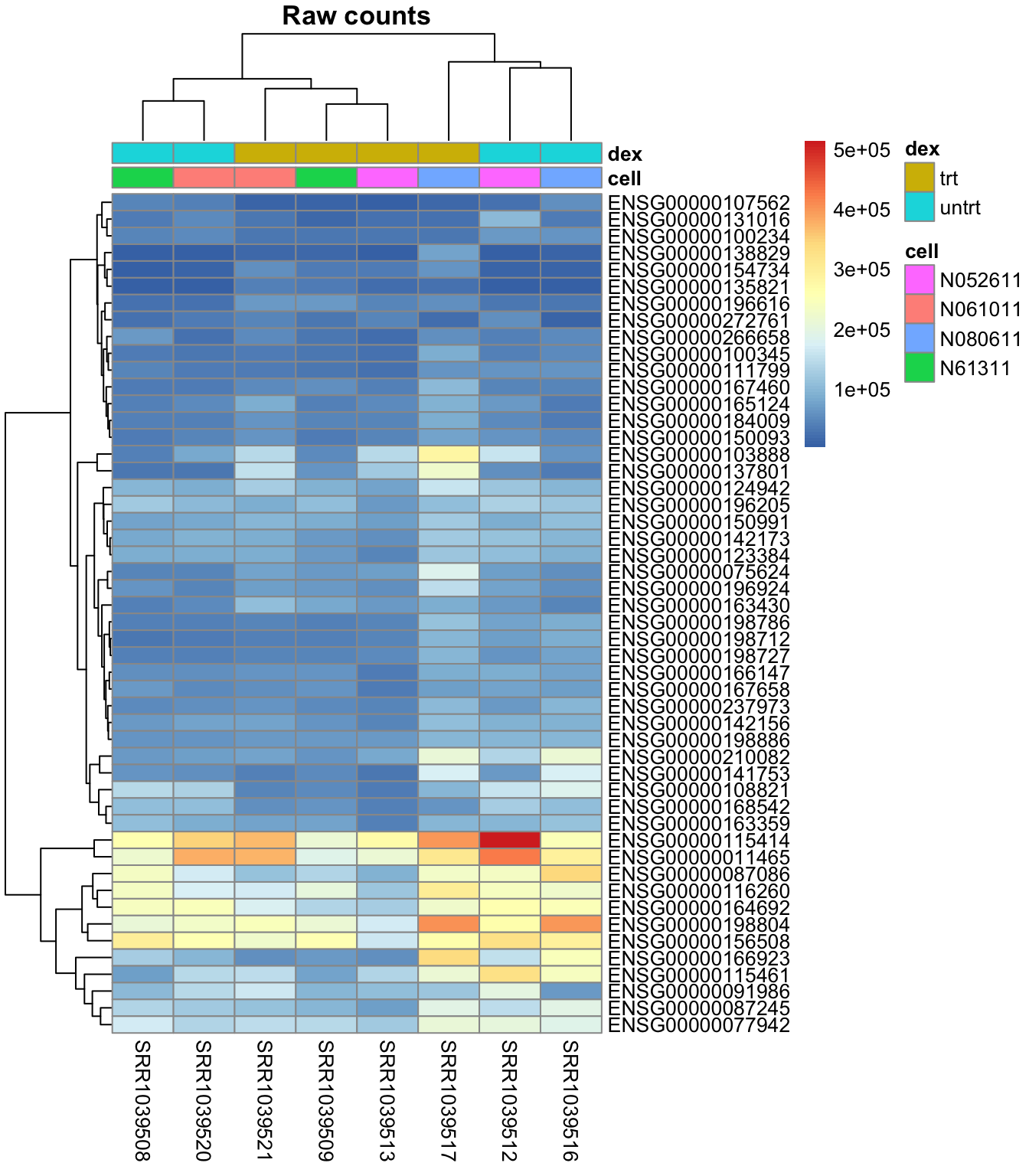
Plot a heatmap with the data log2 transformed to get data that are normal distributed:
pheatmap::pheatmap(log2(mat+1),
annotation = select(metadata, cell, dex),
main = "Log2 transformed counts, no scaling")
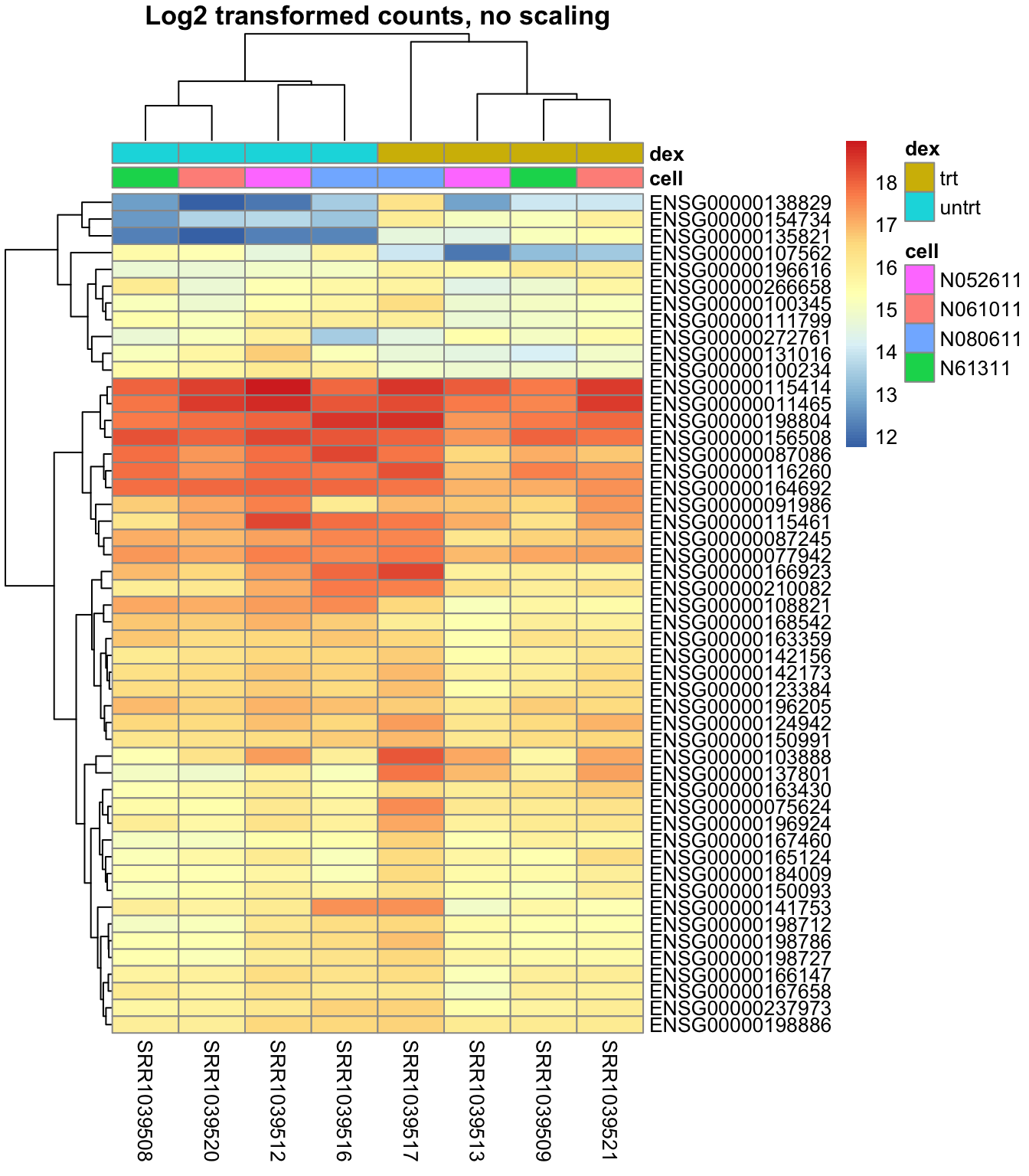
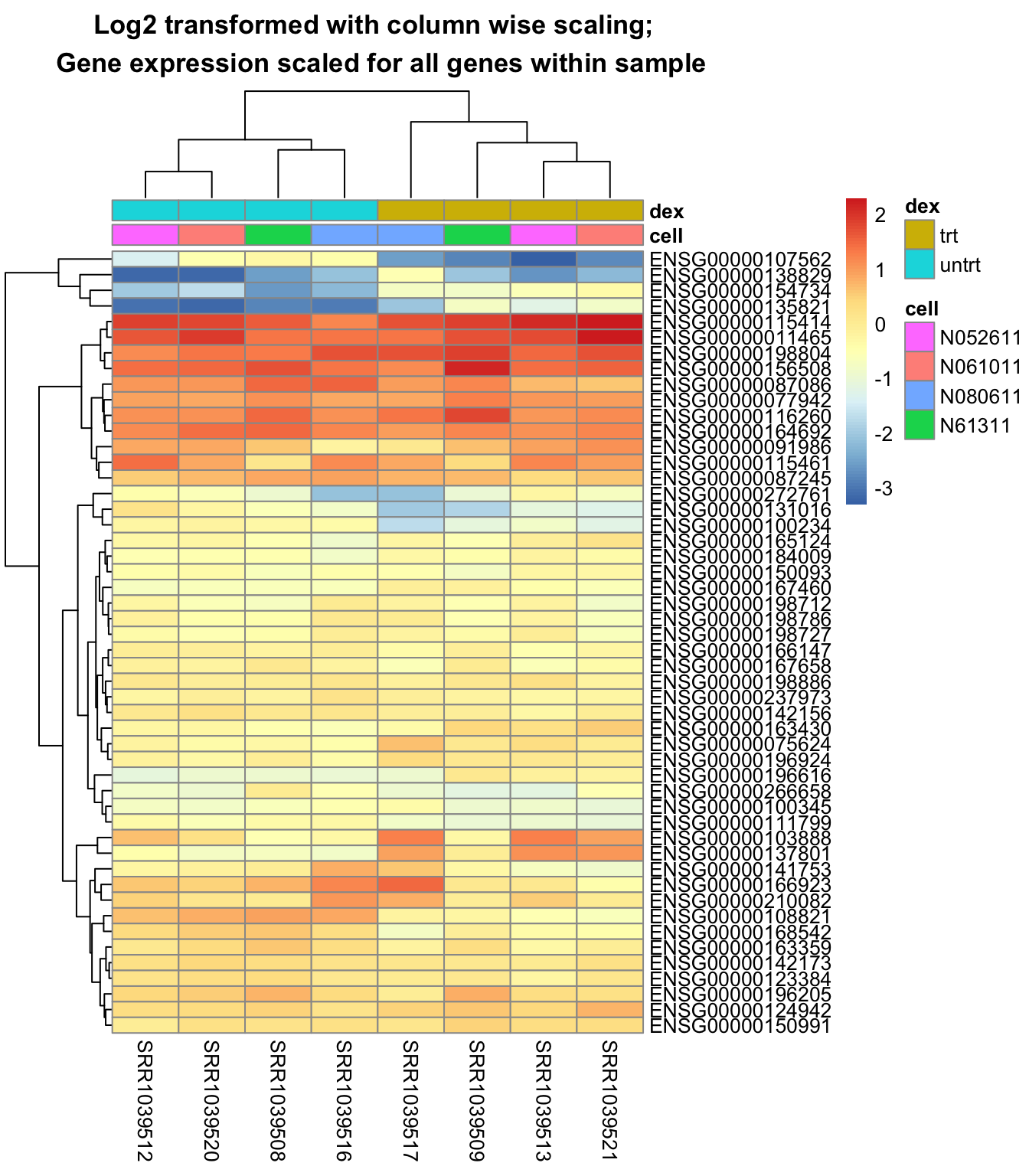
Plot a heatmap with the data log2 transformed, but scaling is performed column wise:
pheatmap::pheatmap(scale(log2(mat+1)),
annotation = select(metadata, cell, dex),
main = "Log2 transformed with column wise scaling; \n Gene expression scaled for all genes within sample")
Plot a heatmap with the data log2 transformed, and properly scaled row-wise:
pheatmap::pheatmap(t(scale(t(log2(mat+1)))),
annotation = select(metadata, cell, dex),
main = "Log2 transformed with row wise scaling; \n Individual gene expression scaled between samples")

The last heatmap show the difference in gene expression between samples, while the second last show the difference of gene expression within each sample. So it is important to do the scaling in the desired direction.
- Posted on:
- June 29, 2022
- Length:
- 5 minute read, 927 words
- Categories:
- R RNAseq Statistics Visualizations
- Tags:
- z-score heatmap scale gene expression
- See Also:
- Read and merge multiple files by folder