How to make R and Python scripts, and make them executable
Or; "How to Calculate %GC content from a fasta.file" --R vs python--
By Synnøve Yndestad in R python sequencing Bioinformatics
August 17, 2021
While practicing my R and Python skills, I have written a script in both R and Python that will perform the same task. Namely take a DNA sequence from a fasta file, count it’s length, and the number of G’s, C’s and N’s while taking upper and lowercase format into account. Then it prints a message with the calculation results. I also included a step that will count how many seconds it takes to do the the calculations, so we can compare which script runs fastest. This post will show you how to create a script from scratch and make it executable on your local computer. I use a MAC, so this tutorial is for any UNIX-like operating systems including Linux.
The first step is obviously to write the script. You can write your code anywhere you like, even notepad will do. Using a Source-code editor will add syntax highlighting, indentation, auto-complete and visualize bracket matching. This will make your life so much easier and colorful. I use RStudio for R (and occasionally python) code, but I also like
Atom when making i.e shell scripts. Atom supports all kinds of encoding and you can toggle between code grammar with two clicks. Reading someone else’s code, or your own after some time, is not always self explanatory. Therefor I try to be overly generous in including explanations for each step using #
# Explanation that will not be included in interpreting the code
# It explains what the code does because
TheCode -> Is_not(c("always", "explaining") %>% itself
Make the file executable in three steps by:
1- Save your file using file extension .R for R scripts and .py for python scripts.
2- Set the address to R/python in the first line using a shebang “#!” and path/to/bin/program.
If you don’t know the address, you can find it by typing “which R” or “which python” in bash terminal.
3- Make it executable with chmod by typing in bash terminal
chmod a+x nameOfFile.R or chmod a+x nameOfFile.py
The chmod command changes the access permissions of the file where:
a = all users
x = execute
then run the program from your terminal in the working directory by typing:
Rscript nameOfFile.R and python nameOfFile.py
Copy paste the code below and give it a try!
Or Download the Scripts and example FASTA files from my
GitHub
The R script
#!/usr/local/bin/r
################################
# File name: GC_calc.R
# R version 4.1.0
# Author: Synnøve Yndestad
# Date: 15.08.2021
# Description:
## This R-script will take a DNA sequence in FASTA format and
## -Compute the length and GC content of a DNA sequence while
## considering both uppercase and lowercase letters.
## -Show processing time.
## -Print output in terminal
################################
# Ask the user for an input and store it as a variable
# get a DNA sequence from a FASTA file:
cat("Please enter name of FASTA file:");
FASTA <- readLines("stdin",n=1);
# Set start time for processing time
start_time <- Sys.time()
# Read FASTA file, save as dna variable
dna = seqinr::read.fasta(FASTA, as.string = TRUE)
# fasta file is read as list, we really just want the first list.
# Subset the first element of the first list:
dna <- dna[[1]]
# Calculate Lenght by counting the number of characters in string
DNAlenght <- nchar(dna)
# Load required packages.
# Tidyverse is a set of packages that work well together including stringr needed for str_count()
# library(tidyverse)
# Count "c" + "C" in dna
no_c <- sum(stringr::str_count(dna, c("c", "C")))
# Count "g" + "G" in dna
no_g <- sum(stringr::str_count(dna,c("g", "G")))
# Count "n" + "N" in dna
no_n <- sum(stringr::str_count(dna,c("n", "N")))
# Calculate GC percentage without N or n bases
gc_percent <- ((no_c+no_g)*100/(DNAlenght-no_n))
# Attach descriptive string to output
OutputString = "%GC content without N bases is:"
# Print name of sequence:
attr(dna, "Annot")
# print output using strings, functions and variables
print(paste("The DNA sequence is", nchar(dna) ,"base pairs long with", no_n ,"number of N bases"), quote = FALSE)
# print gc_percent results rounded to 2 decimals by round()
print(c(OutputString,round(gc_percent,2)), quote = FALSE)
# Save time at end
end_time <- Sys.time()
# Print time elapsed from input
print(paste("---Calculated in", (end_time - start_time), "seconds ---" ), quote = FALSE)
The python script
##! /Users/syndestad/opt/anaconda3/bin/python
"""
File name: GC_calc.py
Python version: 3.8.3
Author: Synnøve Yndestad
Date: 15.08.2021
Description:
This python program will take a DNA sequence in FASTA format as input and
-Compute the length and GC content of a DNA sequence while considering both uppercase and lowercase letters.
-Show processing time.
-Print output in terminal
"""
# Ask for an input file and store it as a variable
#get DNA sequence:
Input_fasta_file = input("Please enter name of FASTA file:")
# import time module
import time
# Set start time for processing time
start_time = time.time()
# Make a function that reads FASTA line by line
def readFASTA(FASTAfile): # Define function and argument
content = [] # Place to put the data
with open(FASTAfile, "r") as file: # read only
for line in file:
line = line.strip() # remove n\ and white spaces
content.append(line) # add each line as it is read
return content
# Use the function to read FASTA
dna = readFASTA(Input_fasta_file)
# Save the first row as input file name
name = dna[0]
# Join all elements to one, exclude first row with name
dna = "".join(dna[1:])
# We now have readable data!
# Count C’s + c's in DNA sequence
no_c=dna.count('c')+dna.count('C')
# Count G’s + c's in DNA sequence
no_g=dna.count('g')+dna.count('G')
# Count N's+n's in DNA sequence
no_n=dna.count('n')+dna.count('N')
# get the length of the DNA sequence
dna_len=len(dna)
# compute gc percentage - N bases
gc_perc=(no_c+no_g)*100.0/(dna_len-no_n)
# Print the name if input sequence
print(name)
# print output using strings, functions and variables.
print("The DNA sequence is", len(dna), "base pairs long with", no_n, "number of N bases")
# Print output text using %% to print a % symbol
# Formatting strings; %%, %-operator separating formatting string and the value to replace the placeholder
# Formatting string example: %5.2f
# % =indicates that a format follows
# 5 = total number of digits
# 2 = number of digits following the .
# f = letter indicating the type of the value to format
# f is fixed point
# Print GC content rounded by 2 decimals
print("The DNA sequence’s GC content is %.2f %%" % gc_perc)
# Print processing time in seconds
print("---Calculated in %s seconds ---" % (time.time() - start_time))
After placing the two scripts in your working directory, go to the same directory in terminal and make it executable with chmod.
The two programs will run when you have both the script-file and fasta file in your working directory. After initializing with Rscript nameOfFile.R or python nameOfFile.py, they will ask for the name of a fasta file. When you enter the name in the terminal and press enter, it will start calculating the output.
Input data
One particularly popular genome at the moment is the SARS-CoV-2 virus. We can examine some of it’s features using our script.
The NCBI virus database keeps growing with more genomes from all over the world added every day. There are manye search options, i.e tax ID, lineages like B.1.617.2
delta variant etc.
Filter yor search, click on the sequence of interest and download some sequences in FASTA format by clicking “send to”, “destination file”, “format fasta”, and put it in your working directory.
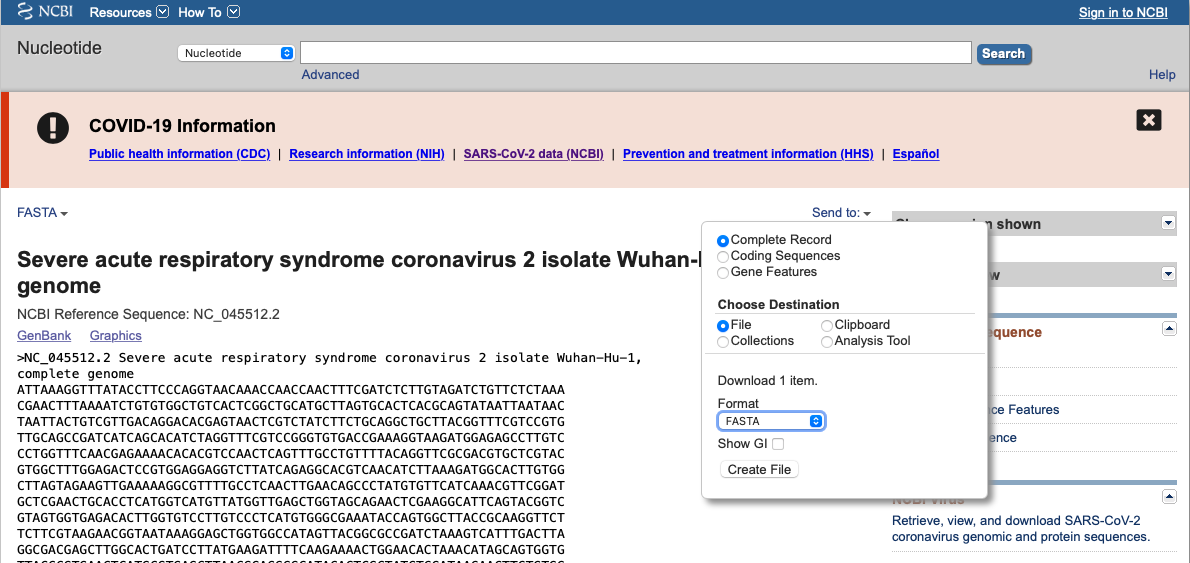
The fasta format is a text format widely used for nucleotide sequences. The first line always starts with a “>” and includes a description of the sequence. The actual sequence starts at line 2. Which is why we started to read from line 2 in the script.
I have downloded some examples saved as:
1 – COVID.sequence.fasta – The reference genome for the novel corona virus SARS-CoV-2, seq no
NC_045512.2
2 – deltaNoN.sequence.fasta – A B.1.617.2 delta variant
MZ714377.1 without any “N”.
3 – deltaWithN.sequence.fasta – A B.1.617.2
OU488266.1 with “N”.
4 – e_coli.sequence.fasta – To include a lareger file, fetch the E.coli genome
NC_000913.3.
How did they perform?
I ran my R and python program on all four sequences, and the output is pasted in below.
Test 1: The SARS-CoV-2 refseq genome:
Rscript GC_calc.R
Please enter name of FASTA file:COVID.sequence.fasta
[1] “>NC_045512.2 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome”
[1] The DNA sequence is 29903 base pairs long with 0 number of N bases
[1] %GC content without N bases is: 37.97
[1] —Calculated in 0.118160963058472 seconds —
python GC_calc.py
Please enter name of FASTA file: COVID.sequence.fasta
NC_045512.2 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome
The DNA sequence is 29903 baseparis long with 0 number of N bases
The DNA sequence’s GC content is 37.97 %
—Calculated in 0.0016603469848632812 seconds —
Test 2: The delta variant with no “N” in sequence
Rscript GC_calc.R
Please enter name of FASTA file: deltaNoN.sequence.fasta
[1] “>MZ714377.1 Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/OH-UHTL-1075/2021, complete genome”
[1] The DNA sequence is 29896 base pairs long with 0 number of N bases
[1] %GC content without N bases is: 37.93
[1] —Calculated in 0.116411924362183 seconds —
python GC_calc.py
Please enter name of FASTA file: deltaNoN.sequence.fasta
MZ714377.1 Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/OH-UHTL-1075/2021, complete genome
The DNA sequence is 29896 baseparis long with 0 number of N bases
The DNA sequence’s GC content is 37.93 %
—Calculated in 0.0008590221405029297 seconds —
Test 3: The delta variant with incomplete bases:
Rscript GC_calc.R
Please enter name of FASTA file: deltaWithN.sequence.fasta
[1] “>OU488266.1 Severe acute respiratory syndrome coronavirus 2 genome assembly, complete genome: monopartite”
[1] The DNA sequence is 29903 base pairs long with 1895 number of N bases
[1] %GC content without N bases is: 38.02
[1] —Calculated in 0.101728916168213 seconds —
python GC_calc.py
Please enter name of FASTA file: deltaWithN.sequence.fasta
OU488266.1 Severe acute respiratory syndrome coronavirus 2 genome assembly, complete genome: monopartite
The DNA sequence is 29903 baseparis long with 1895 number of N bases
The DNA sequence’s GC content is 38.02 %
—Calculated in 0.0016481876373291016 seconds —
Test 4: Lets try something bigger, like E.coli NC_000913:
**Rscript GC_calc.R **
Please enter name of FASTA file: e_coli.sequence.fasta
[1] “>NC_000913.3 Escherichia coli str. K-12 substr. MG1655, complete genome”
[1] The DNA sequence is 4641652 base pairs long with 0 number of N bases
[1] %GC content without N bases is: 50.79
[1] —Calculated in 0.525228977203369 seconds —
python GC_calc.py
Please enter name of FASTA file: e_coli.sequence.fasta
NC_000913.3 Escherichia coli str. K-12 substr. MG1655, complete genome
The DNA sequence is 4641652 baseparis long with 0 number of N bases
The DNA sequence’s GC content is 50.79 %
—Calculated in 0.07539200782775879 seconds —
They both give the same output, and neither takes long to run. Half a second at the longest. As we can see, the E.coli sequence is much longer, and has a GC% of 50.79, vs the SARS-CoV-2 virus GC% of 37.97.
Conclusion:
Both the R and python scrip do the job, but the python script is faster. For bigger data computing time also grows. So if speed is important, python appears to perform better than R. I am more comfortable doing data wrangling and statistics in R, and I am therefor tempted to use R for everything. This got me thinking of the saying; “If all you have is a hammer, everything looks like a nail”. Looking beyond your familiar tools when encountering new problems will also bring new solutions. New is not always better, but we can always try.
- Posted on:
- August 17, 2021
- Length:
- 9 minute read, 1914 words
- Categories:
- R python sequencing Bioinformatics
- Tags:
- DNA programming fasta