NGS sequencing file formats
By Synnøve Yndestad in Bioinformatics sequencing R bash
November 14, 2021
Sequencing data comes in a wide variety of formats and they contain very specific information. This is a collection of notes on different formats, and how to interact with some of them using command line tools like samtools, or R.
Next Generation Sequencing (NGS) technology in brief:
NGS and Sanger sequencing is similar in principle. DNA polymerase adds fluorescent nucleotides to a growing DNA template strand. DNA bases are identified when each base C, T, G, A, emits a fluorescent signal as they are added to a nucleic acid chain.
The sequencing machine is in essence a very fancy camera, and it takes “time-lapse” images of the growing DNA clusters on the flow cell, processing millions of fragments simultaneously. Hence the term “massively parallel sequencing”. In contrast, Sanger sequencing only sequence one fragment at a time.
Before sequencing with Illumina, DNA is first fragmented into short sequences. Samples are then tagged by adding adapters and bar codes, creating sequencing libraries. When we provide a unique bar code to each sample, we can pool multiple samples to the same flow cell, and later extract the captured sequences by index from the pooled soup of sequences.
One fragment is considered to be one read, and when we are talking about read depth it is how many times the area has been covered by read fragments. So when we have a read depth of 100, it means that there are 100 read fragments that has been aligned to that area of the genome.
Depending on your type of project, different bioinformatic workflows may be appropriate. Tools also change over time, but a general illustrative workflow to identify variants/mutations is provided in the image below.
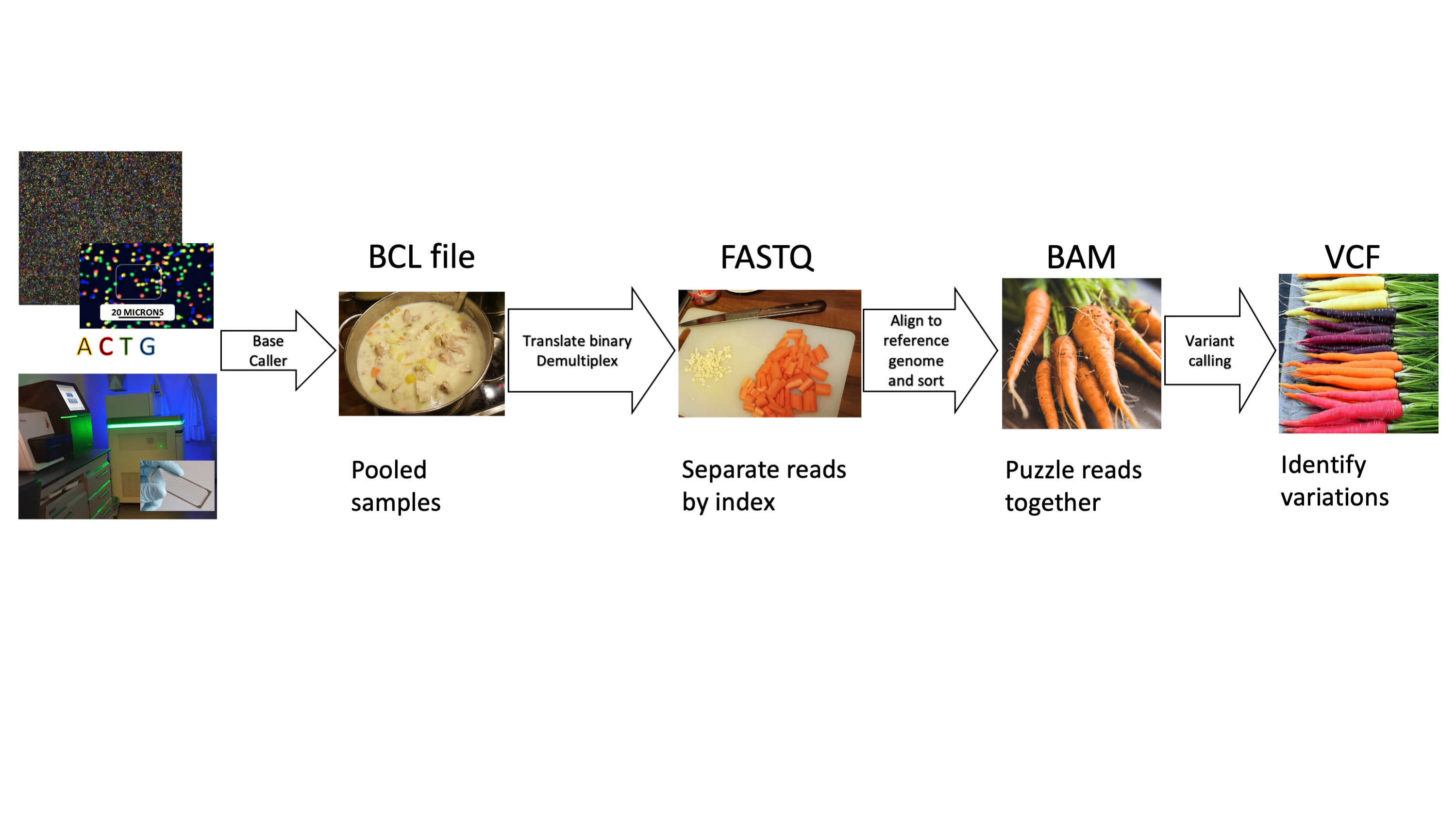
SAM BAM, BAI, BED, GTF, GFF… A brief introduction on most formats can be found here: https://genome.ucsc.edu/FAQ/FAQformat.html
A note on coordinates, 0-based vs 1-based:
The SAM, VCF, GFF and Wiggle formats are using the 1-based coordinate system. The BAM, BCFv2, BED, and PSL formats are using the 0-based coordinate system.
1-based coordinate system: A coordinate system where the first base of a sequence is one. In this co-ordinate system, a region is specified by a closed interval. For example, the region between the 3rd and the 7th bases inclusive is [3, 7]. The SAM, VCF, GFF and Wiggle formats are using the 1-based coordinate system.
0-based coordinate system: A coordinate system where the first base of a sequence is zero. In this coordinate system, a region is specified by a half-closed-half-open interval. For example, the region between the 3rd and the 7th bases inclusive is [2, 7). The BAM, BCFv2, BED, and PSL formats are using the 0-based coordinate system.
BCL files
The Illumina sequencers produce Binary Base Call (BCL) files. The raw data files contains the base and the quality score, which is the confidence of the call. This happens in real time for each cycle in the sequencing run and is produced by the Real-Time Analysis Software (RTA). The sequencer adds information about the location on the flow cell, what tile and lane.
In short, the BCL file contains base calls and quality scores for each tile for each cycle in binary format.
BCL files are converted to FASTQ files by tools like bcl2fasq and bclconvert.
FASTQ files
FASTQ files are text files, and thus readable from the terminal. They contain a number of entries where each entry has 4 lines.
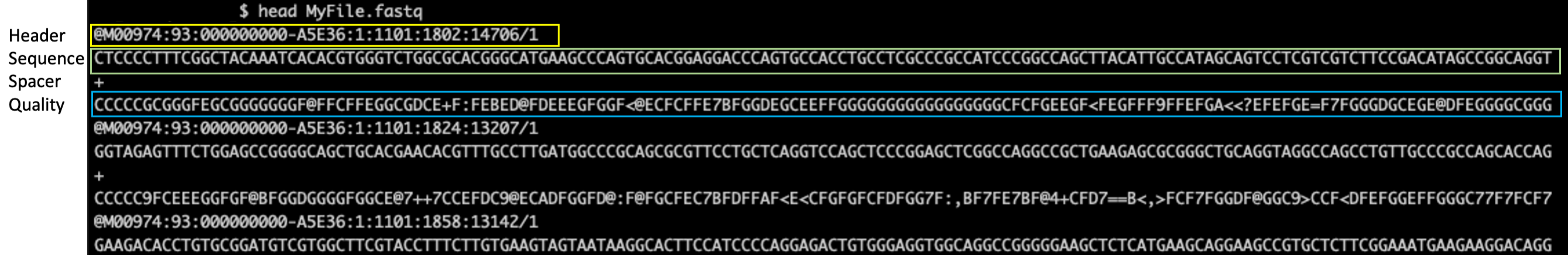
1- Header. Starts with @.
@<instrument>:<run number>:<flowcell ID>:<lane>:<tile>:<x-pos>:<y-pos> <read>:<is filtered>:<control number>:<sample number>
2- The sequence
3- A spacer/seperator “+” indicating start of Quality score
4- Base call quality scores. Phred +33 encoded (ASCII)
To interpret the quality score, see Illumina help pages.
Paired end reads are stored in two separate files.
This means, that to count the number of reads in a FASTQ file from the terminal is quite straight forward. Just count the number of lines in the file, and divide by 4.
Count lines with wc -l:
myFile.fastq wc -l
Now include some calculations. Count lines and divide by 4
echo $(cat myFile.fastq |wc -l)/4|bc
Most likely, the file is compressed to save space, then the file has a .gz extension. Then you need to add an unzip command.
In Linux, the zcat command allows us to view a compressed file without uncompressing and changing the source file.
Uncompress, count lines and divide by 4:
echo $(zcat myFile.fastq.gz|wc -l)/4|bc
In MacOS zcat appends a .Z to the output file, which creates a “can’t stat” error. So, when on a Mac I use gunzip with the -c command to uncompress the file to stdout without changing the original file. Using only gunzip without the -c will delete the original file.
Uncompress, count lines and divide by 4:
echo $(gunzip -c myFile.fastq.gz|wc -l)/4|bc
The output is the number of reads in the FASTQ file.
Viewing FASTQ files with ShortRead in R
FASTQ files can be viewed and manipulated using the bioconductor R package ShortRead :
This package implements sampling, iteration, and input of FASTQ files. The package includes functions for filtering and trimming reads, and for generating a quality assessment report. Data are represented as DNAStringSet-derived objects, and easily manipulated for a diversity of purposes. The package also contains legacy support for early single-end, ungapped alignment formats.`
library(ShortRead)
Get an example file from the ShortRead package:
fastqFilepath <- system.file(package="ShortRead", "extdata", "E-MTAB-1147","ERR127302_1_subset.fastq.gz")
fastqFile <- readFastq(fastqFilepath)
fastqFile
## class: ShortReadQ
## length: 20000 reads; width: 72 cycles
Now we have an R element of class ShortReadQ containing
Subset the first 5
fastqFile[1:5]
## class: ShortReadQ
## length: 5 reads; width: 72 cycles
Look at the first 5 reads
head(sread(fastqFile), 5)
## DNAStringSet object of length 5:
## width seq
## [1] 72 GTCTGCTGTATCTGTGTCGGCTGTCTCGCGGGAC...GTCAATGAAGGCCTGGAATGTCACTACCCCCAG
## [2] 72 CTAGGGCAATCTTTGCAGCAATGAATGCCAATGG...CAGTGGCTTTTGAGGCCAGAGCAGACCTTCGGG
## [3] 72 TGGGCTGTTCCTTCTCACTGTGGCCTGACTAAAA...TGGCATTAAGAAAGAGTCACGTTTCCCAAGTCT
## [4] 72 CTCATCCACACCTTTGGTCTTGATGGCTGTTTCA...CAAAGCATCCCGCTCAGCATCAAAGTTAGTATA
## [5] 72 GTTTGGATATATGGAGGATGGGGATTATTGCTAG...GGATGGATAGTAATAGGGCAAGGACGCCTCCTA
Look at quality scores of the first 5 reads
head(quality(fastqFile), 5)
## class: FastqQuality
## quality:
## BStringSet object of length 5:
## width seq
## [1] 72 HHHHHHHHHHHHHHHHHHHHEBDBB?B:BBGG<D...ABFEFBDBD@DDECEE3>:?;@@@>?=BAB?##
## [2] 72 IIIIHIIIGIIIIIIIHIIIIEGBGHIIIIHGII...IIIHIIIHIIIIIGIIIEGIIGBGE@DDGGGIG
## [3] 72 GGHBHGBGGGHHHHDHHHHHHHHHFGHHHHHHHH...HHHHHHHHGHFHHHHHHHHHHHHHH8AGDGGG>
## [4] 72 IIIIIIIIIIIIIIIIIHIIIGHIIIIIIIIIII...HIIIIIIIIIIHIIIIHIIHIGDIIHIHIIIHF
## [5] 72 GGEGGE8EEEDDBD?EEEDEGGG3GE<EEEBBEE...DC;;CB8BBB?E>E?CGEGG<G@F>GGB>D###
Translate quality encoding:
encoding(quality(fastqFile))
## ! " # $ % & ' ( ) * + , - . / 0 1 2 3 4 5 6 7 8 9 :
## 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25
## ; < = > ? @ A B C D E F G H I J K L M N O P Q R S T
## 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51
## U V W X Y Z [ \\ ] ^ _ ` a b c d e f g h i j k l m n
## 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77
## o p q r s t u v w x y z { | } ~
## 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93
Get the translated quality score, selecting the first 5 sequences and the first 20 bases:
as(quality(fastqFile), "matrix")[1:5,1:20]
## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10] [,11] [,12] [,13] [,14]
## [1,] 39 39 39 39 39 39 39 39 39 39 39 39 39 39
## [2,] 40 40 40 40 39 40 40 40 38 40 40 40 40 40
## [3,] 38 38 39 33 39 38 33 38 38 38 39 39 39 39
## [4,] 40 40 40 40 40 40 40 40 40 40 40 40 40 40
## [5,] 38 38 36 38 38 36 23 36 36 36 35 35 33 35
## [,15] [,16] [,17] [,18] [,19] [,20]
## [1,] 39 39 39 39 39 39
## [2,] 40 40 39 40 40 40
## [3,] 35 39 39 39 39 39
## [4,] 40 40 40 39 40 40
## [5,] 30 36 36 36 35 36
Get the ID of the first 5 sequences
id(fastqFile)[1:5]
## BStringSet object of length 5:
## width seq
## [1] 53 ERR127302.8493430 HWI-EAS350_0441:1:34:16191:2123#0/1
## [2] 53 ERR127302.21406531 HWI-EAS350_0441:1:88:9330:2587#0/1
## [3] 55 ERR127302.22173106 HWI-EAS350_0441:1:91:10434:14757#0/1
## [4] 54 ERR127302.10402268 HWI-EAS350_0441:1:42:13757:9559#0/1
## [5] 54 ERR127302.19486260 HWI-EAS350_0441:1:80:13838:5544#0/1
See the ShortRead vignette for more functionality.
Alignment files, BAM/SAM
When we get a sequence from an Illumina sequencing machine, it is just a short piece of ACTG characters around 200bp long. To put the sequence into a context, we can use an aligner i.e the Burrows-Wheeler Aligner (BWA) to align a sequence to a reference. Alignment files such as SAM og BAM files can be inspected in the Integrative Genomic Viewer (IGV).
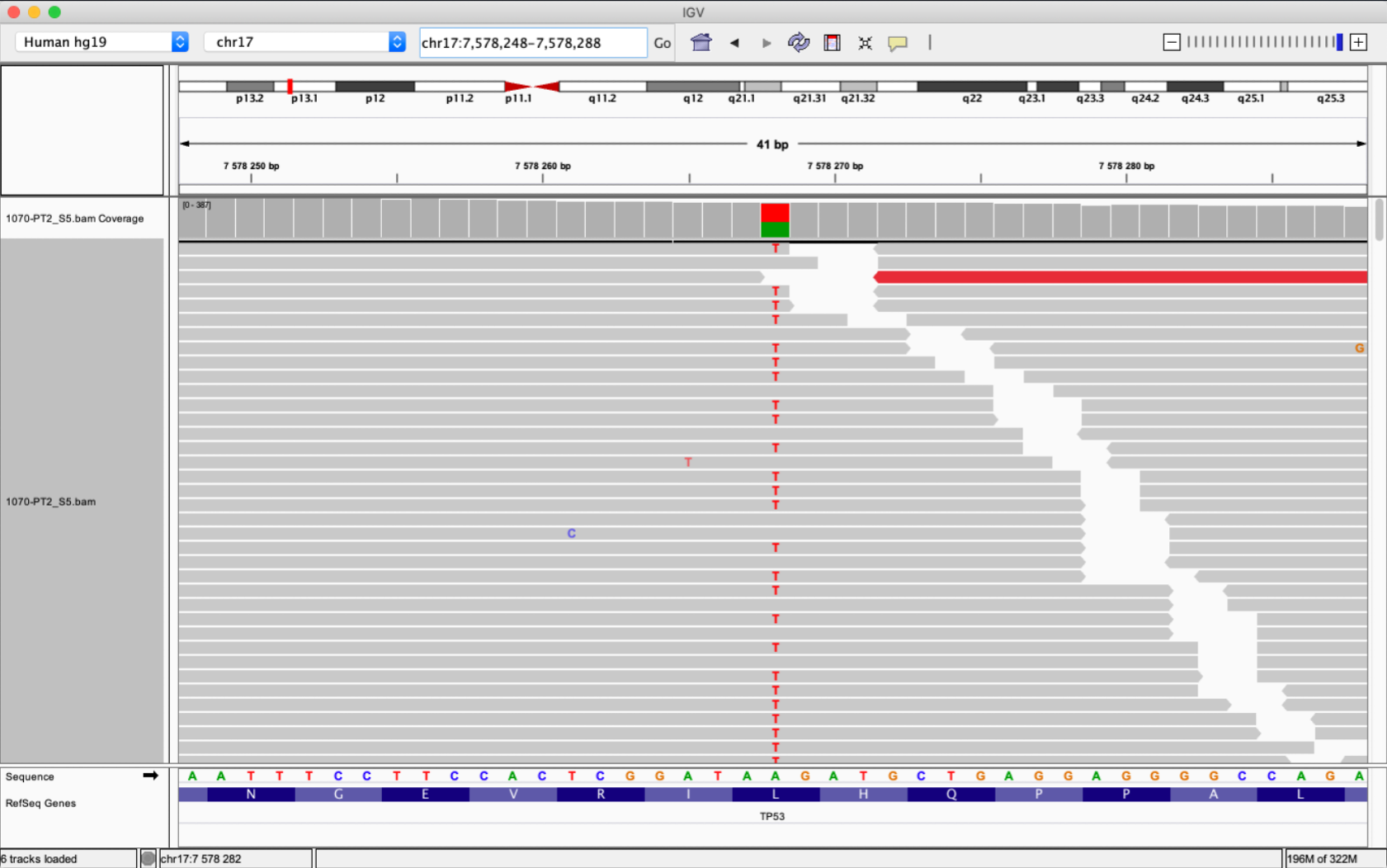
BAM is a compressed binary version of the Sequence Alignment/MAP (SAM) file format.
https://samtools.github.io/hts-specs/SAMv1.pdf
“It is a TAB-delimited text format consisting of a header section, which is optional, and an alignment section. If present, the header must be prior to the alignments. Header lines start with ‘@’, while alignment lines do not. Each alignment line has 11 mandatory fields for essential alignment information such as mapping position, and variable number of optional fields for flexible or aligner specific information.”
The anatomy of a BAM file can look quite intimidating, and it it is very heavy in information, in addition to size.
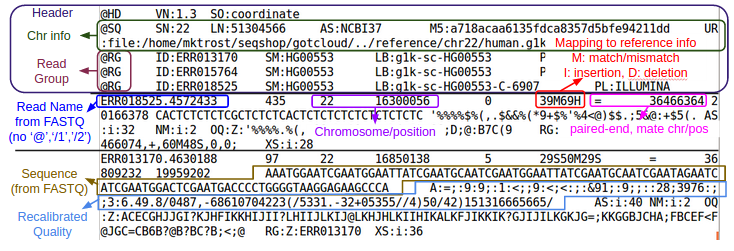
In brief from the https://samtools.github.io/hts-specs/SAMv1.pdf specs:
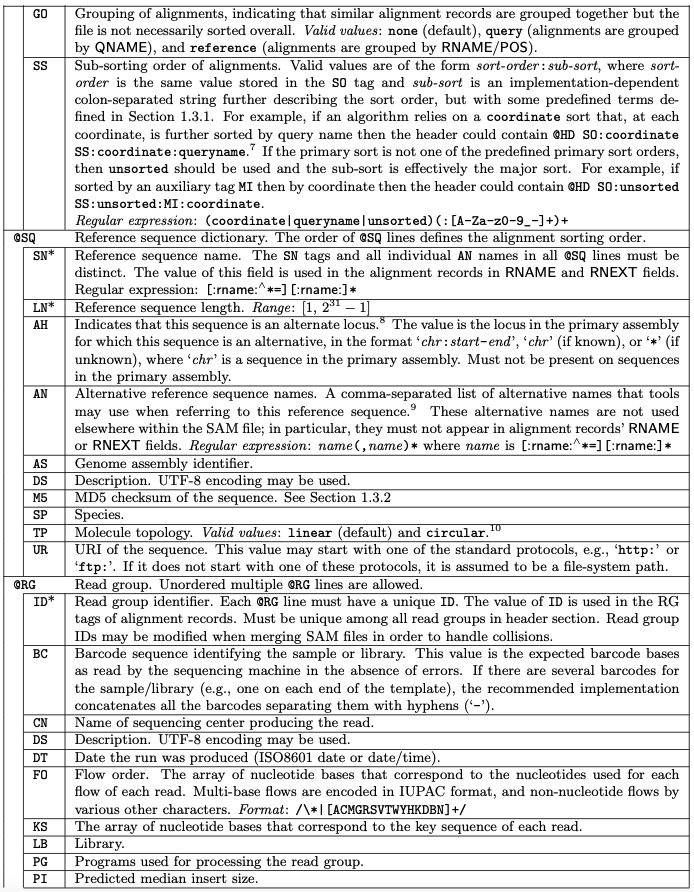
All lines in Header section starts with a @
The header section, can be long, with the following tags:


The Alignment section
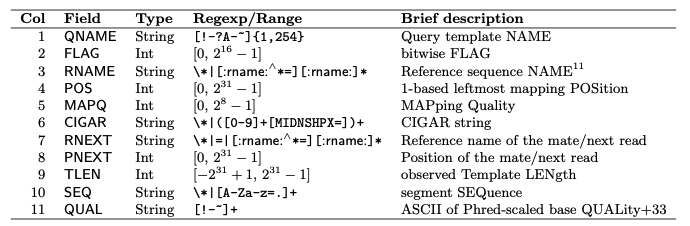
“In the SAM format, each alignment line typically represents the linear alignment of a segment. Each line consists of 11 or more TAB-separated fields. The first eleven fields are always present and in the order shown below; if the information represented by any of these fields is unavailable, that field’s value will be a placeholder, either ‘0’ or ‘*’ as determined by the field’s type. The following table gives an overview of these mandatory fields in the SAM format:”
Concise Idiosyncratic Gapped Alignment Report, or CIGAR
The CIGAR string values:

The CIGAR string gives you a lot of information about an alignment to a reference. It tells you which bases aligns and the positions of those that don´t when there are deletions or insertions.
Consider the following alignment:
Reference Position: 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
Reference Sequence: T C C G A G T A A T C A G G T C G G T T
My_read : G A G A A A A C A G G T C
My_read alignment start at RefSeq position 4, and the corresponding CIGAR string 3M1D2M1D7M indicate that the alignment has 3 Matches, 1 Deletion, 2 Matches, 1 Insertion and 7 Matches.
Working with bam files in command line (bash or UNIX) using samtools
SAMtools http://samtools.sourceforge.net!
“Samtools is a set of utilities that manipulate alignments in the SAM (Sequence Alignment/Map), BAM, and CRAM formats. It converts between the formats, does sorting, merging and indexing, and can retrieve reads in any regions swiftly.”
Since BAM files are in binary code, we can not look at their content directly in the terminal by
head mydata.bam
In stead we can use SAMtools! Type samtools view to get all the options in samtools. ‘view’ in samtools converts the binary code into a readable format.
Use samtools view with -H argument to view the BAM file header.
samtools view -H mydata.bam
## @HD VN:1.3 SO:coordinate
## @SQ SN:chrM LN:16571
## @SQ SN:chr1 LN:249250621
## @SQ SN:chr2 LN:243199373
## @SQ SN:chr3 LN:198022430
## @SQ SN:chr4 LN:191154276
## @SQ SN:chr5 LN:180915260
## @SQ SN:chr6 LN:171115067
## @SQ SN:chr7 LN:159138663
## @SQ SN:chr8 LN:146364022
## @SQ SN:chr9 LN:141213431
## @SQ SN:chr10 LN:135534747
## @SQ SN:chr11 LN:135006516
## @SQ SN:chr12 LN:133851895
## @SQ SN:chr13 LN:115169878
## @SQ SN:chr14 LN:107349540
## @SQ SN:chr15 LN:102531392
## @SQ SN:chr16 LN:90354753
## @SQ SN:chr17 LN:81195210
## @SQ SN:chr18 LN:78077248
## @SQ SN:chr19 LN:59128983
## @SQ SN:chr20 LN:63025520
## @SQ SN:chr21 LN:48129895
## @SQ SN:chr22 LN:51304566
## @SQ SN:chrX LN:155270560
## @SQ SN:chrY LN:59373566
## @RG ID:SampleID-017t PL:ILLUMINA SM:SampleID-017t
## @PG ID:bwa PN:bwa VN:0.7.7-isis-1.0.2
When FASTQ files are aligned, the output is not automatically sorted and the aligment output is in random order. Many tools require that the bam file is ordered according to the position /coordinate in the reference genome, and may be important for downstream applications. If the file is not sorted you can sort it by:
samtools sort sample.bam -o sample.sorted.bam
When SO:coordinate is present in the header, the file is sorted.
Now, look at the first 5 sequences in the bam file.
samtools view mydata.bam | head -n5
## M06980:28:000000000-J96WN:1:1107:11059:14650 97 chrM 1 32 13S63M chr21 11011484 0 AGACATCACGATGGATCACAGGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCATGCATTTGGTATTTTCGT CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGGGEGGGGGFGGGGGGGEGFGGFGGGGC NM:i:0 MD:Z:63 AS:i:63 XS:i:53 RG:Z:SampleID-017t
## M06980:28:000000000-J96WN:1:1118:16906:7981 161 chrM 1 32 13S63M chr21 11011484 0 AGACATCACGATGGATCACAGGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCATGCATTTGGTATTTTCGT CCCCCGGGGGDGGGGGGGGGGGGGGGFGGGGFF<FGGGGGCGGGGFCFFGGG<FGF8FFGGDD<CEEFFGFGGGGE NM:i:0 MD:Z:63 AS:i:63 XS:i:53 RG:Z:SampleID-017t
## M06980:28:000000000-J96WN:1:1102:22111:9884 99 chrM 1 9 37S39M = 1 66 TAGCCCACACGTTCCCCTTAAATAAGACATCACGATGGATCACAGGTCTATCACCCTATTAACCACTCACGGGAGC CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG NM:i:0 MD:Z:39 AS:i:39 XS:i:52 RG:Z:SampleID-017t
## M06980:28:000000000-J96WN:1:2106:18941:5154 163 chrM 1 60 3S72M = 34 109 ATGGATCACAGGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCATGCATTTGGTATTTTCGACTGGGGGGT 9B<CC-EFCFEAFEFF<AFFC@8@F<FEEGF@F8<FGGGGDGEEGDDC,<FEC99C9CFECEFFG,CCCGGC@7C NM:i:1 MD:Z:62T9 AS:i:67 XS:i:43 RG:Z:SampleID-017t
## M06980:28:000000000-J96WN:1:2110:3587:13276 99 chrM 1 3 30S46M = 1 64 CACGTTCCCCTTAAATAAGACATCACGATGGATCACAGGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCAT CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGEGGGGGGG NM:i:0 MD:Z:46 AS:i:46 XS:i:56 RG:Z:SampleID-017t
To find how many aligments we have in a bam file, we can do a word count on lines:
samtools view mydata.bam | wc -l
## 9906610
Or use samtools flagstat:
samtools flagstat mydata.bam
## 9906610 + 0 in total (QC-passed reads + QC-failed reads)
## 952542 + 0 secondary
## 0 + 0 supplementary
## 1091510 + 0 duplicates
## 9793399 + 0 mapped (98.86% : N/A)
## 8954068 + 0 paired in sequencing
## 4477034 + 0 read1
## 4477034 + 0 read2
## 5684672 + 0 properly paired (63.49% : N/A)
## 8788734 + 0 with itself and mate mapped
## 52123 + 0 singletons (0.58% : N/A)
## 2896038 + 0 with mate mapped to a different chr
## 2675441 + 0 with mate mapped to a different chr (mapQ>=5)
To find how many sequences there are in the bam file and how long they are, we can look at the header using the -H flag
samtools view -H mydata.bam | grep "SN:"
echo ' '
# Count the sequences
echo 'Number of Sequences in bam file:'
echo ' '
samtools view -H mydata.bam | grep -c "SN:"
## @SQ SN:chrM LN:16571
## @SQ SN:chr1 LN:249250621
## @SQ SN:chr2 LN:243199373
## @SQ SN:chr3 LN:198022430
## @SQ SN:chr4 LN:191154276
## @SQ SN:chr5 LN:180915260
## @SQ SN:chr6 LN:171115067
## @SQ SN:chr7 LN:159138663
## @SQ SN:chr8 LN:146364022
## @SQ SN:chr9 LN:141213431
## @SQ SN:chr10 LN:135534747
## @SQ SN:chr11 LN:135006516
## @SQ SN:chr12 LN:133851895
## @SQ SN:chr13 LN:115169878
## @SQ SN:chr14 LN:107349540
## @SQ SN:chr15 LN:102531392
## @SQ SN:chr16 LN:90354753
## @SQ SN:chr17 LN:81195210
## @SQ SN:chr18 LN:78077248
## @SQ SN:chr19 LN:59128983
## @SQ SN:chr20 LN:63025520
## @SQ SN:chr21 LN:48129895
## @SQ SN:chr22 LN:51304566
## @SQ SN:chrX LN:155270560
## @SQ SN:chrY LN:59373566
##
## Number of Sequences in bam file:
##
## 25
The “LN:” number will represent the sequence length.
Now we can use the CIGAR strings in field 6 to extract and count deletions and insertions:
# count the deletions
echo 'Number of deletions:'
samtools view mydata.bam | cut -f6 | grep -c 'D'
# count the insertions
echo 'Number of insertions:'
samtools view mydata.bam | cut -f6 | grep -c 'I'
# count both insertions and deletions
echo 'Number of insertions and deletions:'
# Count either deletions or insertions:
samtools view mydata.bam | cut -f6 | grep -c 'I\|D'
## Number of deletions:
## 114105
## Number of insertions:
## 87048
## Number of insertions and deletions:
## 197796
In field 7 of the bam file we find the RNEXT strings.
From the https://samtools.github.io/hts-specs/SAMv1.pdf manual;
RNEXT: Reference sequence name of the primary alignment of the NEXT read in the template. For the last read, the next read is the first read in the template. If @SQ header lines are present, RNEXT (if not ‘*’ or ‘=') must be present in one of the SQ-SN tag. This field is set as ‘*’ when the information is unavailable, and set as ‘=’ if RNEXT is identical RNAME. If not ‘=’ and the next read in the template has one primary mapping (see also bit 0x100 in FLAG), this field is identical to RNAME at the primary line of the next read. If RNEXT is ‘*’, no assumptions can be made on PNEXT and bit 0x20.
In other words; Alignments with unmapped mate has the label ‘*’
Alignments with mate mapped to same chromosome has the label ‘=’
Count Alignments with unmapped mate and Alignments with mate mapped to same chromosome:
echo 'Number of alignments with unmapped mate:'
samtools view mydata.bam | cut -f7 | grep -c '*'
echo 'Number of alignments with mate mapped to same chromosome:'
samtools view mydata.bam | cut -f7 | grep -c '='
## Number of alignments with unmapped mate:
## 61088
## Number of alignments with mate mapped to same chromosome:
## 6480914
Subset a BAM file by Chromosomal range:
When you need to split the file into smaller pieces for various reasons (interest, increased computing speed, space limitations) we can subset a bam file into multiple files
To specify a bam output, you will need a ‘-b’ flag. Include ‘-h’ to retain header in output.
samtools view -b -h input.bam "Chr17:start-end" > output.bam
I want to subset my file to only chr17.
First, we need to index the bam file and create the bam.bai index file:
samtools index mydata.bam
Now that we have created the index file mydata.bam.bai, we can split it:
samtools view -b mydata.bam chr17 > mydataChr17.bam
Now, lets check the size
echo 'Alignments in original file:'
samtools view mydata.bam | wc -l
echo 'Aligments in subsetted file:'
samtools view mydataChr17.bam | wc -l
## Alignments in original file:
## 9906610
## Aligments in subsetted file:
## 593264
VCF files
The Variant Call Format (VCF) stores the location and type of variant deviating from the reference genome.
The header starts with # and contains various metadata i.e what reference genome was used. The body has 8 mandatory columns, but can contain multiple others as well.
From the wikipedia entry of VCF files:

Looking at vcf file content
Smaller VCF files can be opened in Excel. But just because you can, does not mean that you should.
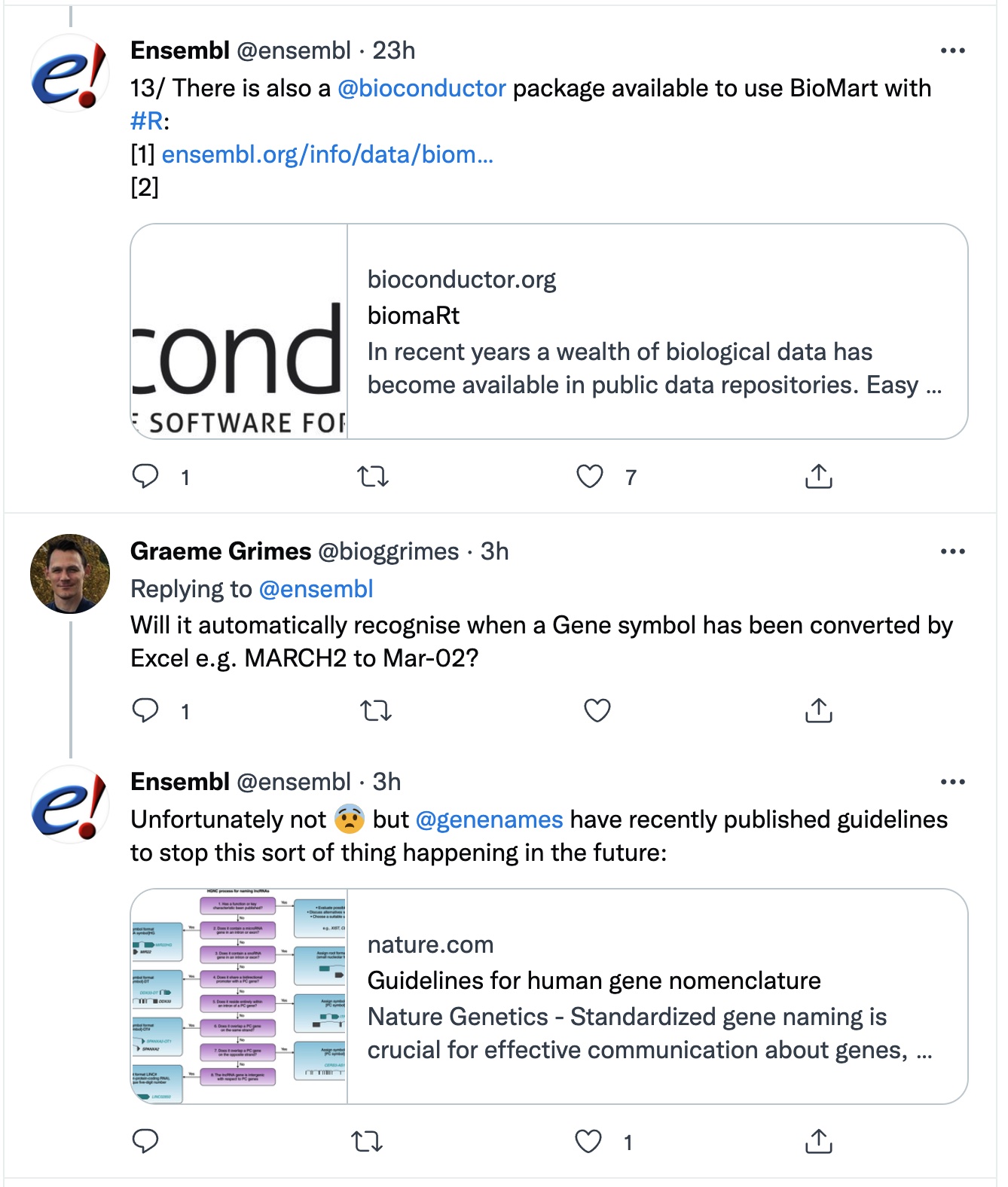 (Twitter 2021-11-12)
(Twitter 2021-11-12)
One tool for working with VCF files in R is vcfR
For simple tasks, like extracting specific information from a VCF file, it is quickly done from the command line
Check out the top 5, exclude header lines starting with #
cat MCF7_S6.vcf | grep -v '^#' | head -n5
## chr1 3638674 . C T 100.00 PASS DP=307 GT:GQ:AD:VF:NL:SB:GQX 1/1💯0,307:1.0000:20:-100.0000:100
## chr1 11205058 . C T 100.00 PASS DP=156 GT:GQ:AD:VF:NL:SB:GQX 1/1💯0,156:1.0000:20:-100.0000:100
## chr1 11288758 . G A 100.00 PASS DP=107 GT:GQ:AD:VF:NL:SB:GQX 1/1💯0,107:1.0000:20:-100.0000:100
## chr1 11301714 . A G 100.00 PASS DP=90 GT:GQ:AD:VF:NL:SB:GQX 1/1💯0,90:1.0000:20:-100.0000:100
## chr1 27023007 . AGGC A 23.00 LowGQ DP=33 GT:GQ:AD:VF:NL:SB:GQX 0/1:23:30,3:0.0909:20:-10.7392:23
How many variants are reported?
cat MCF7_S6.vcf | grep -v '^#' | wc -l
## 728
How many passes filter?
cat MCF7_S6.vcf | grep -v '^#' | cut -f7 | grep 'PASS' | wc -l
## 517
How many Chr3 in file?
grep all but exclude lines starting with #, cut by field code, grep ‘Chr3’, do word count on lines
cat MCF7_S6.vcf | grep -v '^#' | cut -f1 | grep 'chr3' | wc -l
## 90
How many entries are reported for position 186276394 on Chr1?
cat MCF7_S6.vcf | grep -v '^#' | cut -f1,2 | grep 'chr1' | cut -f2 | grep '^186276394$' | wc -l
## 2
FASTA files
FASTA files are text files. They always starts with a header “>” followed by an unique sequence identifier. The sequence is the line after a header line. Multiple sequences can be added.
Sequence1 TCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGA Sequence2 CTCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGA Sequence3 GTCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGATCGA
To count the number of sequences or look at the names, we can fetch the lines containing the sequence identifier “>” and count them:
# Count sequences
echo "Number of sequences in file"
cat MyFile.fasta | grep ">" | wc -l
# List name of sequences
echo "Sequence names are:"
cat MyFile.fasta | grep ">"
## Number of sequences in file
## 3
## Sequence names are:
## >Sequence1
## >Sequence2
## >Sequence3
BED
BED = Browser Extensible Data format
A file format for genomic coordinates with associated annotations. Using coordinates in stead of sequences will decrease computing time, and is easier to manipulate.
Minimum 3 columns with the header
#chr start end
chr1 11167541 11167557
chr1 11168238 11168343
chr1 11169347 11169427
chr1 11169706 11169786
chr1 11172909 11172974
GTF
GTF = Genomic Transfer Format
(GFF = General Feature Format, and GTF = GFF version 2)
A tab delimited file format for gene annotations based on the general feature format.
From genecode :

SAMtools mpileup format
Tab seperated text file, where each line represent a pileup of reds at a single genomic region. Each line represent the position along the genome where a read has aligned. There are multiple fields.
Field 1 - Chromosome number
Field 2 - The position on the chromosome (1-based)
Field 3 - The reference base
Field 4 - Depth. Number of reads at that position
Field 5 - Character string with info on matches, mismatches, indels, strand, mapping quality and starts and ends of reads.
Field 6 - ASCII encoded base quality
Field 7 - ASCII encoded Alignment mapping qualities
Misc
The Human Reference Genome ( GRCh38 - hg38)
- Posted on:
- November 14, 2021
- Length:
- 18 minute read, 3623 words
- Categories:
- Bioinformatics sequencing R bash